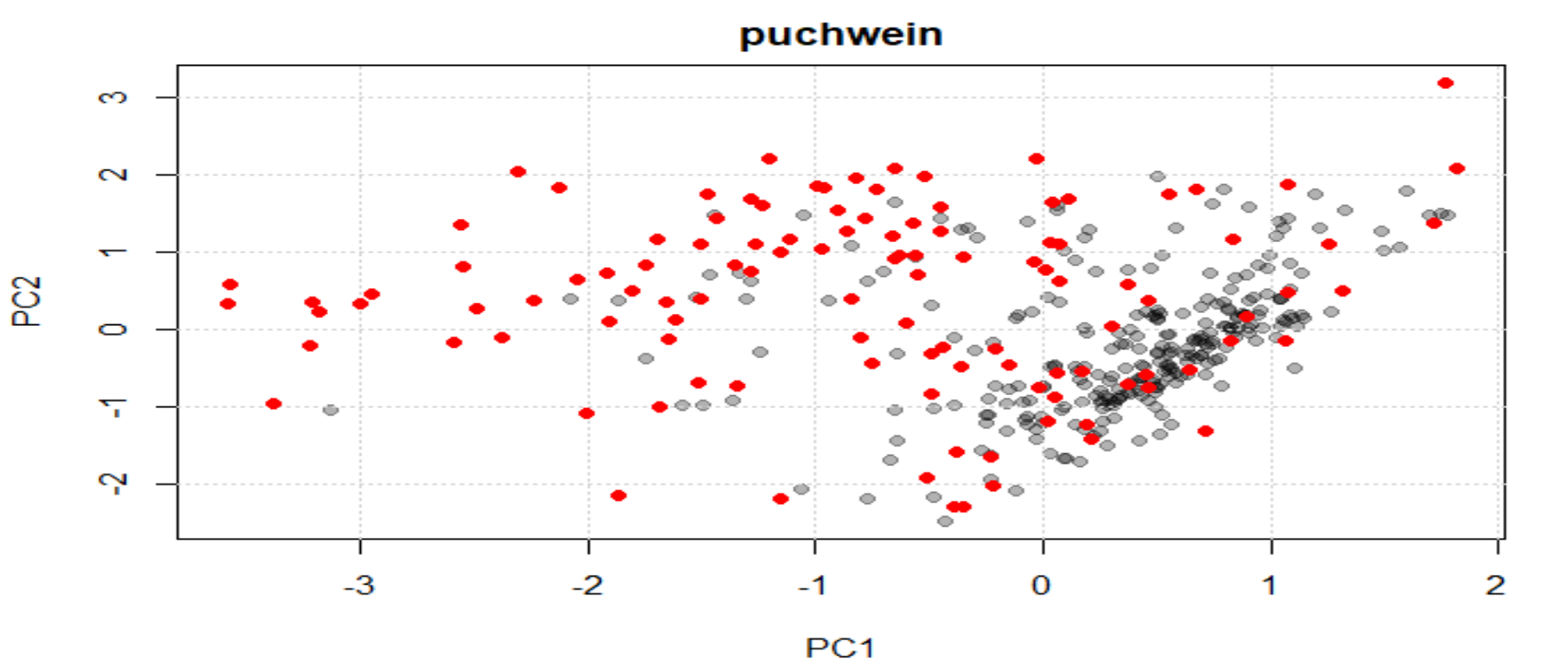
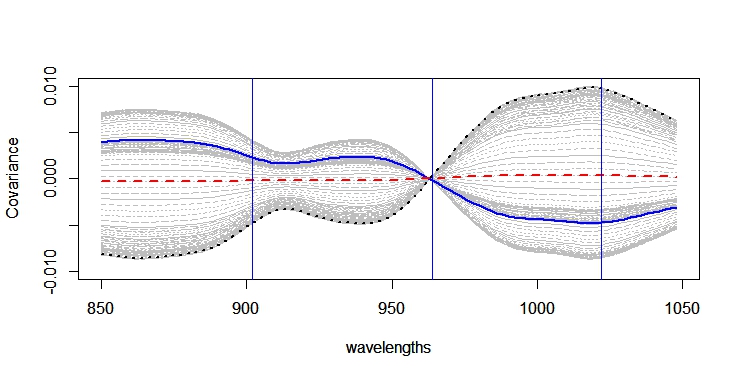
As we saw in the post "Try to find high content gypsum samples (part 3)" , when we develop the principal component analysis as much variance as possible (we determine the explained variance limit) is explained, and unless we saw all the sores maps, we may have not a good idea about what is happening to our data.
The average spectrum represents all the groups, so we cannot expect that with the Mahalanobis distance all the samples with gypsum will be marked as outliers, because the average spectrum represents as well those samples. Only the ones with high and very high gypsum content can probably be marked as outliers by Mahalanobis distance. Now that we have make two samples sets ("gypsum content" and "non or low gypsum content"), we can check the sample sets separately to understand better our data.
Let´s check again the Mahalanobis distance plot once we have, by spectra visualization and discrimination by correlation, the two sample sets ("No" and "Yes" gypsum content):

See how some of the gypsum content samples are over 3 Mahalanobis cut-off (but not all).
Now that the "No" samples are in a new sample set, we can see their spectra:


Let´s see finally the new Mahalanobis distances: