###### LOADING TECATOR DATA ###################################
library(caret)
library(pls)
data(tecator)
#' Loading the tecator data we load two matrices:
#' The spectra matrix "absorp" (raw spectra)
#' We want to create another matrix with MSC math treatment
absorpMSC<-msc(absorp)
#' The constituents matrix "endpoints (Moisture, Fat & Protein)
set.seed(930)
#We can add names to the columns with the wavelengths values.
wavelengths<-as.matrix(seq(850,1048,by=2))
colnames(absorp)<-wavelengths
colnames(endpoints)<- c("Moisture","Fat","Protein")
#' We will model the protein content data and create a data partition
#' leaving 3/4 for the training set and 1/ for the validation set.
#' With the createDataPartition we generate a selection of sample positions
#' in a ramdon order to take after this samples out from the absorp and
#' endpoint matrices.
###### SPLITTING THE DATA #####################################
trainMeats <- createDataPartition(endpoints[,3], p = 3/4)
#'Now we select the correspondant training and validation matrices
#'with the raw and MSC treated spectra
absorpTrain <- absorp[trainMeats[[1]], ]
absorpTrainMSC<-as.matrix(absorpMSC[trainMeats[[1]], ])
absorpTest <- absorp[-trainMeats[[1]], ]
absorpTestMSC <- as.matrix(absorpMSC[-trainMeats[[1]], ])
######### RAW SCAN SPECTRA ##################################################
matplot(wavelengths,t(absorpTrain),type="l",
xlab="wavelengths",ylab="Absorbance",col="blue")
par(new=TRUE)
matplot(wavelengths,t(absorpTest),type="l",
xlab="",ylab="",col="green")
######### MSC SCAN SPECTRA ##################################################
matplot(wavelengths,t(absorpTrainMSC),type="l",xlab="wavelengths",
ylab="transmitance",ylim =c(min(absorpTrainMSC)-0.1,
max(absorpTrainMSC)+0.1),
col="blue")
par(new=TRUE)
matplot(wavelengths,t(absorpTestMSC),type="l",xlab="wavelengths",
ylab="transmitance",ylim =c(min(absorpTrainMSC)-0.1,
max(absorpTrainMSC)+0.1),
col="green")
#'and from the endpoint matrix for every constituent
moistureTrain <- endpoints[trainMeats[[1]], 1]
fatTrain <- endpoints[trainMeats[[1]], 2]
proteinTrain <- endpoints[trainMeats[[1]], 3]
# The rest of the samples go to the Validation Set
moistureTest <- endpoints[-trainMeats[[1]],1]
fatTest <- endpoints[-trainMeats[[1]], 2]
proteinTest <- endpoints[-trainMeats[[1]], 3]
#We can combine these two matrices:
# For Protein
trainDataProt<-cbind(proteinTrain,absorpTrain) #Protein Raw Training
testDataProt<-cbind(proteinTest,absorpTest) #Protein Raw Test
trainDataProtMSC<-cbind(proteinTrain,absorpTrainMSC) #Protein Raw Training
testDataProtMSC<-cbind(proteinTest,absorpTestMSC) #Protein Raw Test
#For Fat
trainDataFat<-cbind(fatTrain,absorpTrain) #Fat Raw Training
testDataFat<-cbind(fatTest,absorpTest) #Fat Raw Test
trainDataFatMSC<-cbind(fatTrain,absorpTrainMSC) #Fat MSC Training
testDataFatMSC<-cbind(fatTest,absorpTestMSC) #Fat MSC Test
#For Moisture
trainDataMoi<-cbind(moistureTrain,absorpTrain) #Moisture Raw Training
testDataMoi<-cbind(moistureTest,absorpTest) #Moisture Raw Test
trainDataMoiMSC<-cbind(moistureTrain,absorpTrainMSC) #Moisture MSC Training
testDataMoiMSC<-cbind(moistureTest,absorpTestMSC) #Moisture MSC Test
##### BUILDING THE MODELS ####################################
##### MODELS FOR MOISTURE
model_moi_raw <- train(moistureTrain~.,data=trainDataMoi, method = "pls",
scale = TRUE,
trControl = trainControl("cv", number = 10),
tuneLength = 20)
model_moi_msc <- train(moistureTrain~.,data=trainDataMoiMSC, method = "pls",
scale = TRUE,
trControl = trainControl("cv", number = 10),
tuneLength = 20)
##### MODELS FOR FAT
model_fat_raw <- train(fatTrain~.,data=trainDataFat, method = "pls",
scale = TRUE,
trControl = trainControl("cv", number = 10),
tuneLength = 20)
model_fat_msc <- train(fatTrain~.,data=trainDataFatMSC, method = "pls",
scale = TRUE,
trControl = trainControl("cv", number = 10),
tuneLength = 20)
##### MODELS FOR PROTEIN
model_prot_raw <- train(proteinTrain~.,data=trainDataProt, method = "pls",
scale = TRUE,
trControl = trainControl("cv", number = 10),
tuneLength = 20)
model_prot_msc <- train(proteinTrain~.,data=trainDataProtMSC, method = "pls",
scale = TRUE,
trControl = trainControl("cv", number = 10),
tuneLength = 20)
###### PREDICTIONS ########################################
## PROTEIN PREDICTIONS
pred_prot_test_raw <- predict(model_prot_raw,testDataProt)
pred_prot_test_msc <- predict(model_prot_msc,testDataProtMSC)
## FAT PREDICTIONS
pred_fat_test_raw <- predict(model_fat_raw,testDataFat)
pred_fat_test_msc <- predict(model_fat_msc,testDataFatMSC)
## MOISTURE PREDICTIONS
pred_moi_test_raw <- predict(model_moi_raw,testDataMoi)
pred_moi_test_msc <- predict(model_moi_msc,testDataMoiMSC)
## PREPARING DATA FOR MONITOR FUNCTION
compare<-cbind(moistureTest,pred_moi_test_raw,pred_moi_test_msc,
fatTest,pred_fat_test_raw,pred_fat_test_msc,
moistureTest,pred_moi_test_raw,pred_moi_test_msc)
ID<-seq(1,52,by=1)
compare<-cbind(ID,compare)
#### MONITORING AND STATISTICS #########################
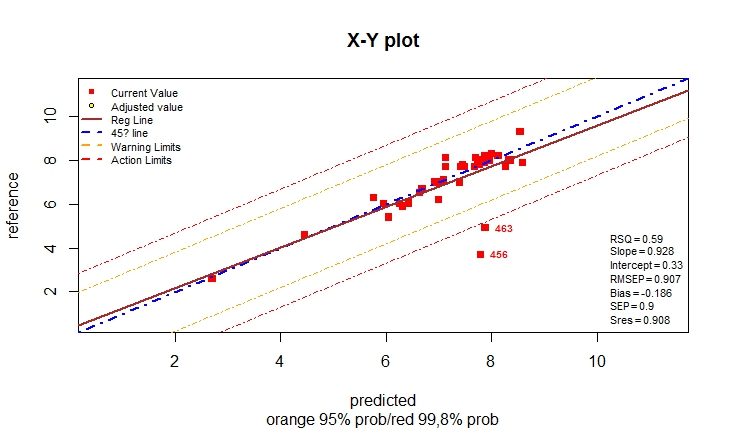
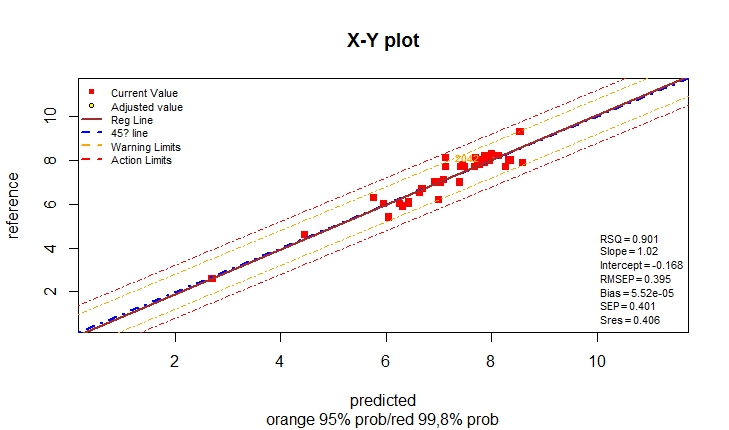
monitor10c26_003(compare[,c(1,2,3)])
monitor10c26_003(compare[,c(1,2,4)])
monitor10c26_003(compare[,c(1,5,6)])
monitor10c26_003(compare[,c(1,5,7)])
monitor10c26_003(compare[,c(1,8,9)])
monitor10c26_003(compare[,c(1,9,10)])
#' For Moisture and Fat there is an improvement using the model
#' with MSC math treatment,
#' For Protein the result are almost the same, but with the
#' raw spectra the is a certain slope and intercept problem,
#' and if corrected there is an improvement in the statistics.