Este espectro en el rango de 400 a 2500 nanómetros, con datos de log(1/R) cada 2 nm,
dispone de 1050 variables, pero lo acotaremos para mayor comodidad al rango de 1100 a
2500 nm, lo que representan 700 variables. En este rango es donde están las bandas de los
analitos de interés (proteína, grasa y humedad). El espectro acotado queda así:
La matriz de datos original consta de 66 filas (equivalen al número de muestras) y de 700
columnas (nº de longitudes de onda). El espectro en negro es el de desviación estándar y nos
da una idea de donde esta la variabilidad en este conjunto de datos.
Ahora vamos a ver el efecto del centrado quitando a cada espectro, el espectro medio:
No están todas las muestras, pero las representadas, nos dan una idea mejor de donde esta
la variabilidad en el espectro, y vemos que coincide con el espectro de desviación
estándar anterior.
Estas muestras se pueden ubicar en un espacio multidimensional de tantas dimensiones
como variables (longitudes de onda), como un punto. La muestra promedio de todas esta
en el centroide de dicho espacio. Como hemos comentado antes, este espacio tiene
correlacionadas las variables entre sí, siendo necesario encontrar nuevas variables no correlacionadas y que sean combinaciones lineales de todas las demás (con mayor o
menor peso). Afortunadamente los programas quimiométricos hacen estas funciones,
quedando en nuestro caso del siguiente modo para los tres primeros componentes
principales de los diez que selecciona:
Podemos elegir las combinaciones que queramos de componentes principales. En este
caso es el PC1, PC2 y PC3, pero podemos seleccionar el 1-2-7, el 1-4-8,....,etc.
Para representar el 100% de la varianza, hubiésemos necesitado las 700 dimensiones, pero
en este caso con diez componentes principales representamos el 99,5%.
Hemos dicho que los componentes principales son combinaciones lineales de las
variables originales y que en el caso del primero representa la mayor fuente de varianza
(en este caso el 82,6%).
Veamos en la siguiente figura como es este primer componente principal:
Se aprecian bandas importantes en entorno a 1940 y 1450 nm (bandas de humedad),
también aparecen otras bandas, de las que hablaremos mas adelante. Todo parece indicar
que este primer componente principal esta representando como parte importante las
bandas de humedad así como de algún otro parámetro.
Comentamos anteriormente que podemos representar estos componentes principales,
los unos respecto a los otros formando planos.
Vamos a mirar el plano formado por el PC1 y PC2:
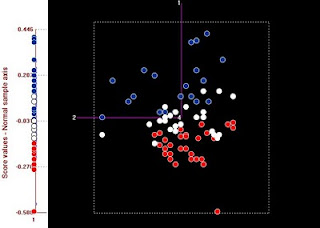
Se han dividido las muestras en 3 tercios acorde a su valor de humedad: Las rojas
representan al tercio con menor humedad, las blancas al intermedio y las azules a las de
mayor humedad. Al proyectar las muestras sobre el eje del PC1, se observa que hay
una gran correlación (0,9) respecto a los valores de humedad.Las proyecciones sobre el
segundo tienen ya muy poca correlación con la humedad.
El primero también tiene cierta correlación con la grasa y la proteína (cerca de 0,3), esto lo
justifican las otras bandas que aparecen en el espectro del primer componente principal,
asociadas a estos dos constituyentes
En los softwares quimiométricos, a partir de la matriz original “X”, creamos dos nuevas
matrices a las que llamaremos: ”P” (loadings o cargas) y “T” (scores o puntuaciones).
Estas dos nuevas matrices combinadas en una determinada fórmula se acercarán en
mayor o menor medida a dar un resultado similar a “X”. Todo dependerá de la matriz de
residuales “E”, que representa a la varianza no explicada.
En el caso de que existan tantos loadings como variables en X, se explicará toda la varianza
(100%), siendo por tanto el residual de cero.
En el caso del software Win ISI, la matriz de loadings es el fichero PCA y la matriz de
scores el fichero LIB. Ambos ficheros deben de estar en la misma carpeta y
conjuntamente representan un cubo entorno a un centroide en el que confluyen los
ejes que representan a los loadings.
Como es lógico solo se podrán representar gráficamente en “3D”, pero podemos hacerlo
con una combinación de diferentes loadings. Un fichero LIB solo lo podremos
representar gráficamente, si en su mismo proyecto está el fichero “PCA”.
El fichero PCA nos aporta los coeficientes de cada longitud de onda respecto a los
distintos componentes principales (PC1, PC2,…..). Por tanto donde se produzcan las
mayores fuentes de varianza, sus longitudes de onda tendrán altos coeficientes en los
primeros componentes principales. En muchas ocasiones estas fuentes de varianza
están relacionadas con características físicas (tamaño de partícula, color, viscosidad,….),
o con determinados analitos incluidos en la muestra y que tienen un amplio rango de
concentración y presentan bandas de absorción en la región NIR (humedad, grasa, proteína,….).
Cada componente principal no representa únicamente a un analito, puede representar a
varios en mayor o menor medida. Para ello es importante fijarse en las los coeficientes en
todas las longitudes de onda.
Puede que la información, en lo que concierne a ciertos analítos, este en los primeros
componentes principales o en los últimos, por lo que decidir sobre el número de componentes principales a seleccionar será uno de los temas a tratar





